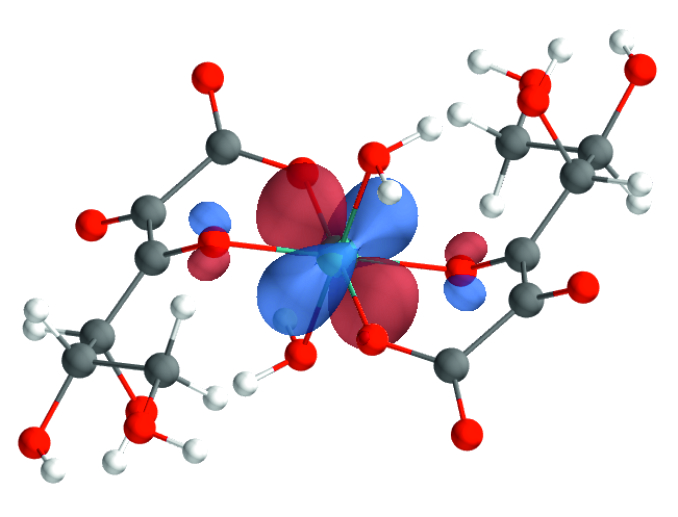
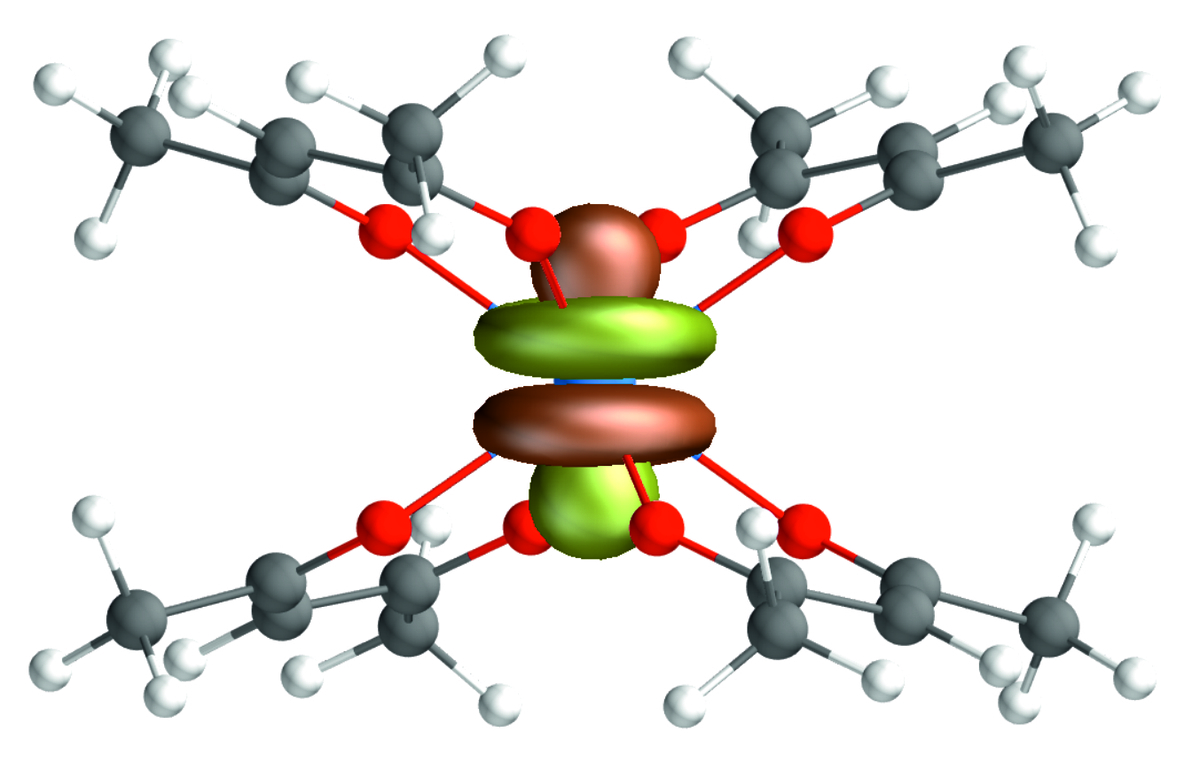
Computational Chemistry
Using ab initio as well as DFT-based methods, the research group "Computational Chemistry" at KIT-INE provides valuable insights on a molecular scale - assisting and complementing experimental investigations in the field of nuclear waste disposal and fundamental radionuclide sciences. There is a wide range of applications for Computational Chemistry at INE: from providing structures of complex chemical systems (including actinides in solution, at surfaces or in solid phases) or thermodynamic data to the reproduction of experimental XAFS spectra. The considered systems vary from molecular species in the gas phase over small complexes in solution to bulk phases or species at mineral/liquid interfaces. New theoretical methods and the constantly improving hardware allow a steady improvement of the description of radionuclide (actinides and fission products) systems at the electronic structure level. These improvements also increase the accuracy and reliability of quantum chemistry as a predictive tool. In particular, theoretical chemistry forms the direct link between basic and applied research in the field of speciation of unknown radionuclides in the environment and in repository-relevant environments (cf., e.g. [1]).
Contact:
Dr. Robert Polly +49 721 608 24396